GeneCodeR spatial signal to test quality of transform
Reload important files recently saved:
library(grid)
library(gridExtra)
library(ggplot2)
main_path <- "~/Documents/main_files/AskExplain/Q4_2022/gcode/"
# Please replace this path
path_to_save <- paste(main_path,"./temp_save_dir/",sep="")
load(file = paste(sep="",path_to_save,"all_genecoder.RData"))Set up the test configuration for GeneCodeR
# Set up genecoder transform information
genecoder.config <- GeneCodeR::extract_config_framework(F)
genecoder.config$transform$from <- 1
genecoder.config$transform$to <- 2
genecoder.config$extract_spots$window_size <- 30Set up validation functions to evaluate statistically significant differences via a t-test, and, cosine similarity.
# Testing functions
# cosine metric for similarity between observations
cosine.simil_sample_and_genes <- function(a,b,non_zero_markers){
return(
list(
sample_wise = do.call('c',parallel::mclapply(c(1:dim(a)[1]),function(X){
lsa::cosine(as.numeric(a[X,non_zero_markers[X,]]),as.numeric(b[X,non_zero_markers[X,]]))
},mc.cores = 8)),
gene_wise = do.call('c',parallel::mclapply(c(1:dim(a)[2]),function(X){
lsa::cosine(as.numeric(a[non_zero_markers[,X],X]),as.numeric(b[non_zero_markers[,X],X]))
},mc.cores = 8))
)
)
}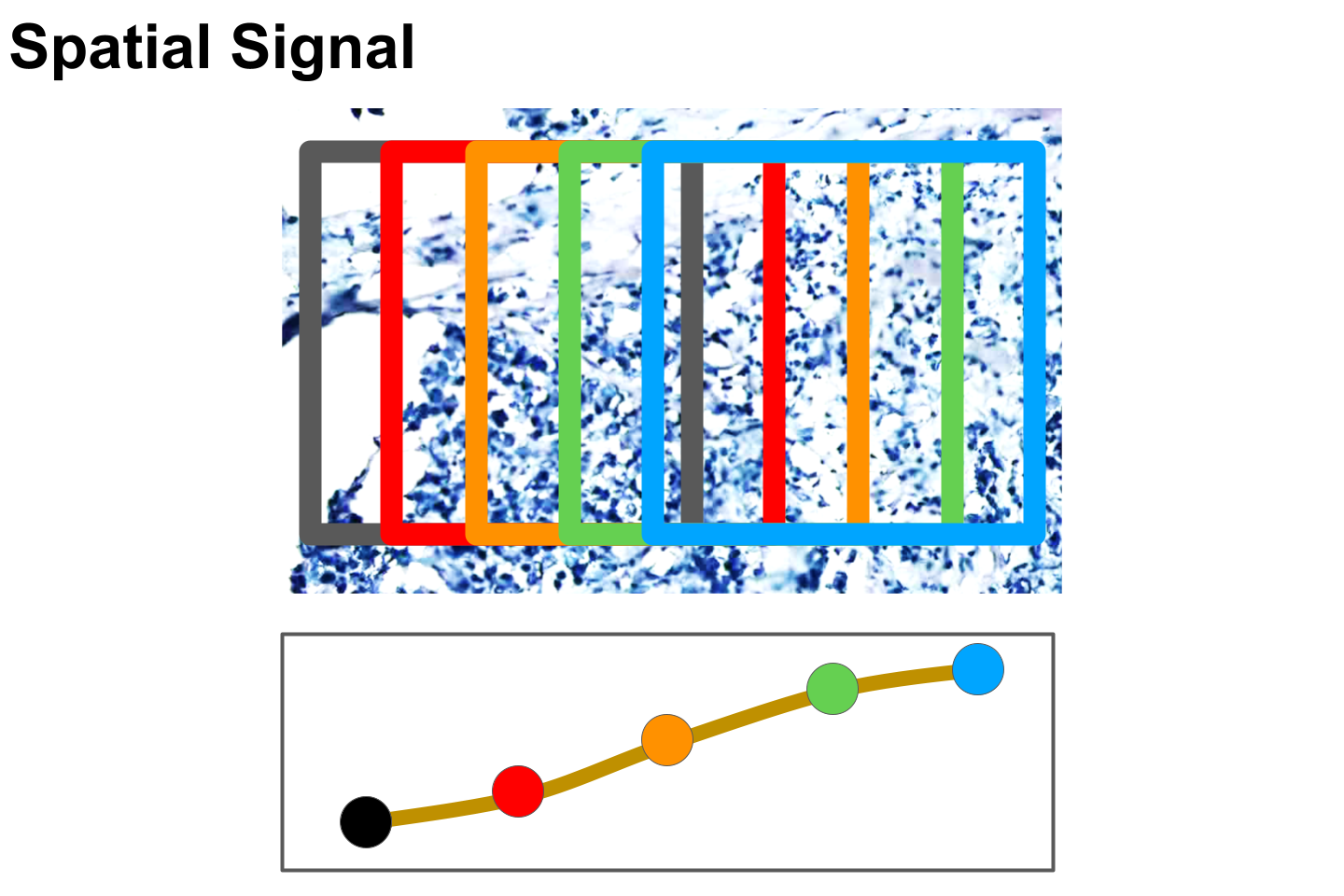
Signal validation
Signal testing is used to evaluate how gene expression signals change over a spatial region when the example image spot slides over a spatial region. It is expected that the gene expression should represent a signal, such that a smoothing model can be learned with a reasonable and flexible fit.
# Spatial signal testing
signal_spot2gex <- list()
for (i in c(1:10)){
genecoder.config$extract_spots$displacement_x <- i*3
genecoder.config$extract_spots$displacement_y <- 0
signal_test_spot_data <- GeneCodeR::prepare_spot(file_path_list = test_file_path_list,meta_info_list = meta_info_list,config = genecoder.config, gex_data = test_gex_data$gex)
signal_spot2gex[[as.character(i)]] <- GeneCodeR::genecoder(model=genecoder.model, x = signal_test_spot_data$spot, config = genecoder.config, model_type = "gcode")
}## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
number_of_top_variable_genes_to_extract_adj.r.squared <- 3
top_genes <- order(apply(base_test_spot_data$gex,2,var),decreasing = T)[1:number_of_top_variable_genes_to_extract_adj.r.squared]
spline_signal_adj.r.squared <- do.call('rbind',lapply(c(top_genes),function(Y){
internal.adj.r2 <- do.call('c',pbmcapply::pbmclapply(c(1:dim(signal_spot2gex[[1]])[1]),function(Z){
signal_spline <- data.frame(gene_level = do.call('c',lapply(c(1:10),function(X){
signal_spot2gex[[X]][Z,Y]
})),displacement=c(1:10))
adj.r.squared <- try(npreg::summary.ss(npreg::ss(x = signal_spline$gene_level,y = signal_spline$displacement))$adj.r.squared,silent = F)
if (!is.character(signal_spline)){
return(as.numeric(adj.r.squared))
}
},mc.cores = 6))
}))
signal_spline <- data.frame(gene_level = do.call('c',lapply(c(1:10),function(X){
signal_spot2gex[[X]][1,top_genes[1]]
})),displacement=c(1:10))
mod.ss <- npreg::summary.ss(npreg::ss(x = signal_spline$gene_level,y = signal_spline$displacement))
plot(signal_spline$gene_level ~ signal_spline$displacement, main = paste("Adj.R-squared: ",mod.ss$adj.r.squared))
lines(signal_spline$gene_level, mod.ss$y, lty = 2, col = 2, lwd = 2)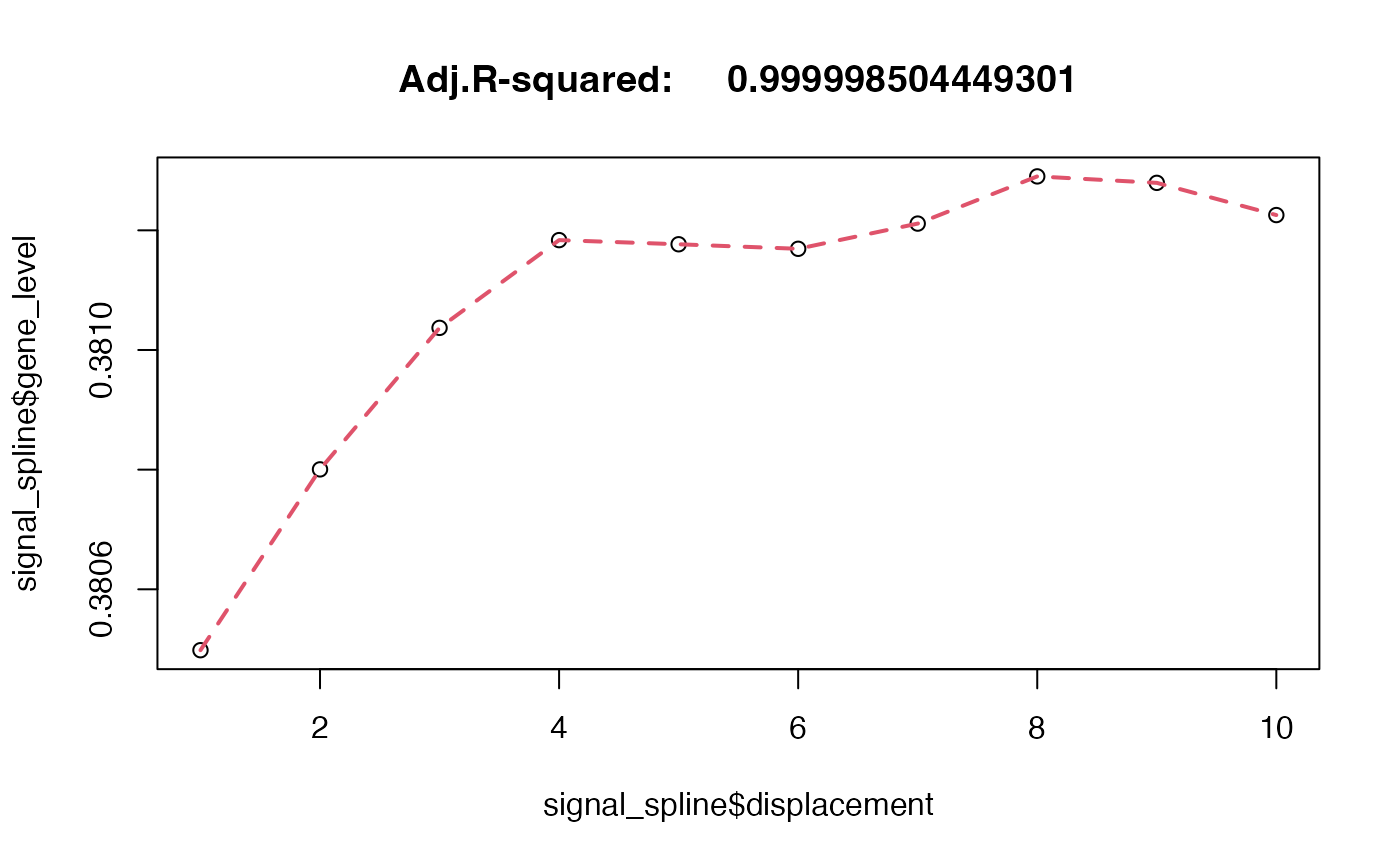
title_name = "spatial_signal"
tissue_name = "breast"
first_most_variable_gene = spline_signal_adj.r.squared[1,]
second_most_variable_gene = spline_signal_adj.r.squared[2,]
third_most_variable_gene = spline_signal_adj.r.squared[3,]
library(ggplot2)
lm <- rbind(c(1,2,3),
c(1,2,3),
c(1,2,3))
g1 <- ggplot(data.frame(Measure=first_most_variable_gene,Metric="Adj.R.Squared of Spline fits \n 1st most variable gene \n across all spatial spots"), aes(x=Metric,y=Measure)) +
geom_violin() + ylim(-1,1)
g2 <- ggplot(data.frame(Measure=second_most_variable_gene,Metric="Adj.R.Squared of Spline fits \n 2nd most variable gene \n across all spatial spots"), aes(x=Metric,y=Measure)) +
geom_violin() + ylim(-1,1)
g3 <- ggplot(data.frame(Measure=third_most_variable_gene,Metric="Adj.R.Squared of Spline fits \n 3rd most variable gene \n across all spatial spots"), aes(x=Metric,y=Measure)) +
geom_violin() + ylim(-1,1)
gg_plots <- list(g1,g2,g3)
final_plots <- arrangeGrob(
grobs = gg_plots,
layout_matrix = lm
)## Warning: Removed 190 rows containing non-finite values
## (`stat_ydensity()`).## Warning: Removed 191 rows containing non-finite values
## (`stat_ydensity()`).## Warning: Removed 190 rows containing non-finite values
## (`stat_ydensity()`).
plot(final_plots)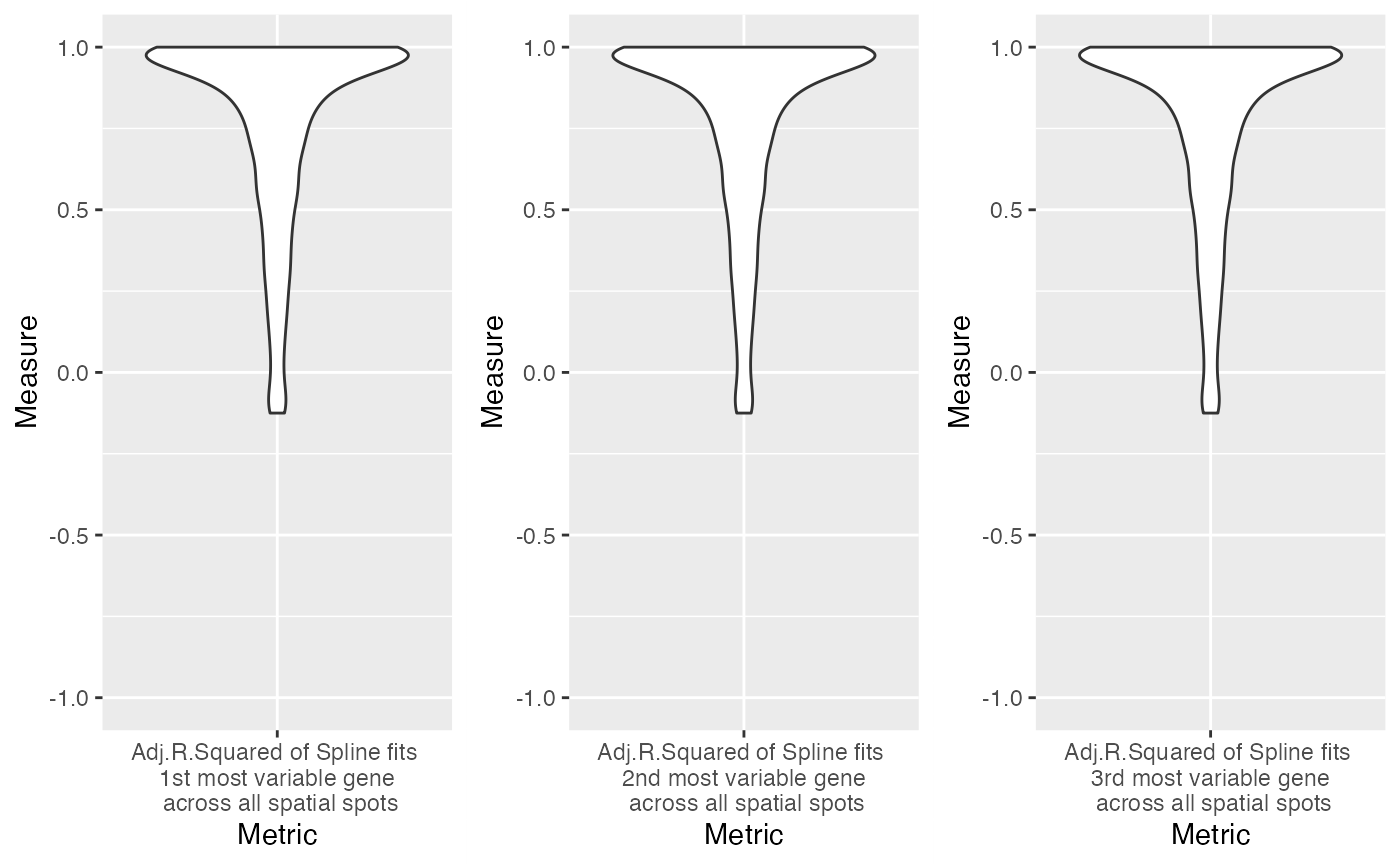
ggsave(final_plots,filename = paste(path_to_save,"/jpeg_accuracy_gcode_",tissue_name,"_SPATIAL/",title_name,"_accuracy_",tissue_name,"_spatial.png",sep=""),width = 7,height=5)
rm(list=ls())
gc()## used (Mb) gc trigger (Mb) limit (Mb) max used (Mb)
## Ncells 1209723 64.7 3366022 179.8 NA 3366022 179.8
## Vcells 2205458 16.9 1553704292 11853.9 16384 1825395395 13926.7