GeneCodeR spatial displacement to test invariance
Reload important files recently saved:
main_path <- "~/Documents/main_files/AskExplain/Q4_2022/gcode/"
# Please replace this path
path_to_save <- paste(main_path,"./temp_save_dir/",sep="")
load(file = paste(sep="",path_to_save,"all_genecoder.RData"))Set up the test configuration for GeneCodeR
# Set up genecoder transform information
genecoder.config <- GeneCodeR::extract_config_framework(F)
genecoder.config$transform$from <- 1
genecoder.config$transform$to <- 2
genecoder.config$extract_spots$window_size <- 30Set up validation functions to evaluate statistically significant differences via a t-test, and, cosine similarity.
# Testing functions
# cosine metric for similarity between observations
test_sample_and_genes <- function(a,b,non_zero_markers,test_type="cosine"){
if (test_type == "t.test"){
return(
list(
sample_wise = do.call('c',parallel::mclapply(c(1:dim(a)[1]),function(X){
t.test(as.numeric(a[X,non_zero_markers[X,]]),as.numeric(b[X,non_zero_markers[X,]]))$p.value
},mc.cores = 8)),
gene_wise = do.call('c',parallel::mclapply(c(1:dim(a)[2]),function(X){
t.test(as.numeric(a[non_zero_markers[,X],X]),as.numeric(b[non_zero_markers[,X],X]))$p.value
},mc.cores = 8))
)
)
}
if (test_type == "cosine"){
return(
list(
sample_wise = do.call('c',parallel::mclapply(c(1:dim(a)[1]),function(X){
lsa::cosine(as.numeric(a[X,non_zero_markers[X,]]),as.numeric(b[X,non_zero_markers[X,]]))
},mc.cores = 8)),
gene_wise = do.call('c',parallel::mclapply(c(1:dim(a)[2]),function(X){
lsa::cosine(as.numeric(a[non_zero_markers[,X],X]),as.numeric(b[non_zero_markers[,X],X]))
},mc.cores = 8))
)
)
}
}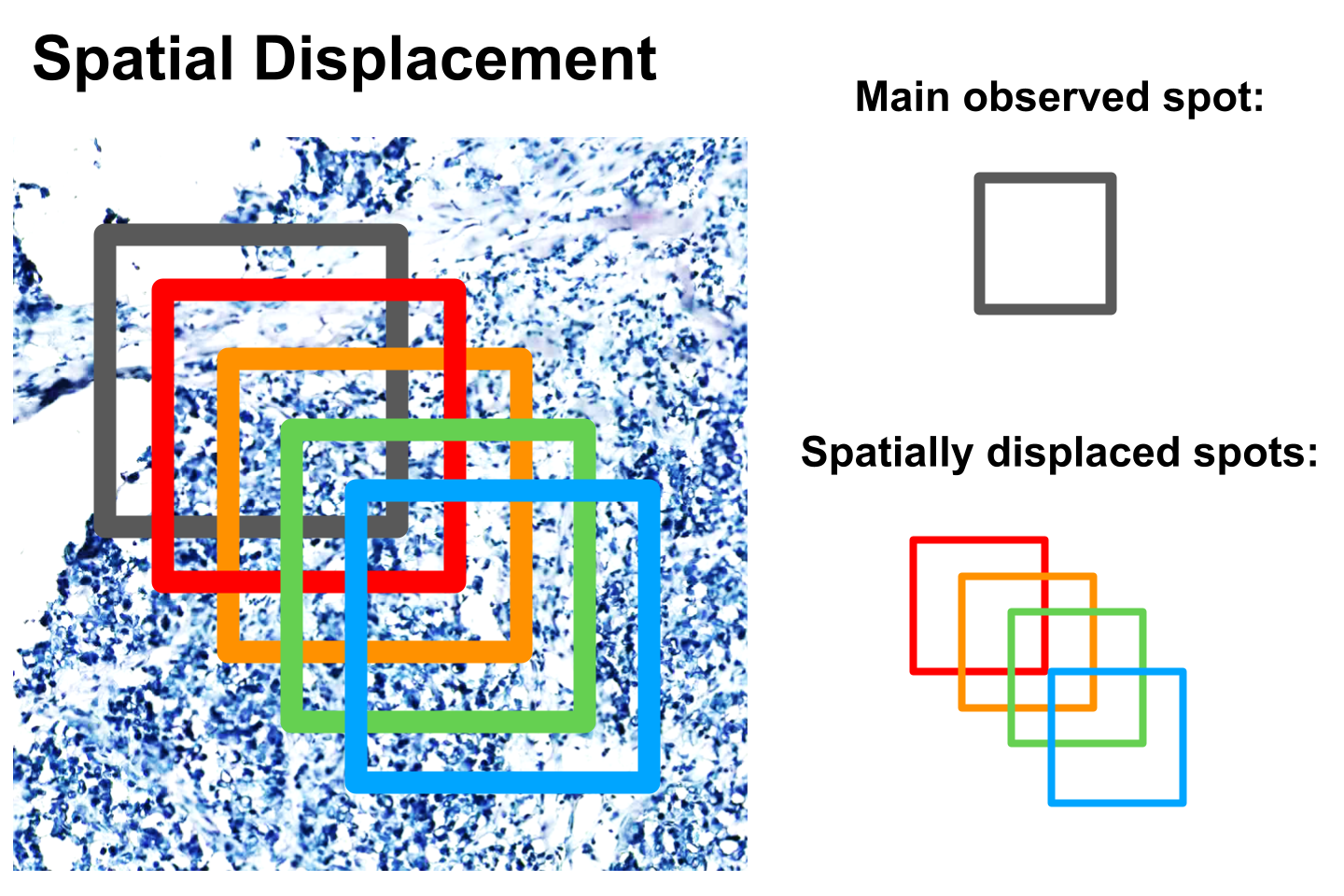
Displacement validation
Displacement testing is used to evaluate how gene levels change when the image spot is displaced from where gene expression is experimentally measured. It is expected that as the displacement increases, the gene expression difference should become weaker in terms of cosine similarity correlation. Notice that from taking gradually increasing displacements, the cosine correlation can be seen to follow a similar trend (e.g. 0 pixel displacement vs 10,20,30 pixel displacements).
# Spatial displacement testing
displace_spot2gex <- list()
for (displace_val in c(0,10,20,30)){
genecoder.config$extract_spots$rotation <- 0
genecoder.config$extract_spots$displacement_x <- displace_val
genecoder.config$extract_spots$displacement_y <- displace_val
displace_test_spot_data <- GeneCodeR::prepare_spot(file_path_list = test_file_path_list,meta_info_list = meta_info_list,config = genecoder.config, gex_data = test_gex_data$gex)
displace_spot2gex[[as.character(displace_val)]] <- GeneCodeR::genecoder(model=genecoder.model, x = displace_test_spot_data$spot, config = genecoder.config, model_type = "gcode")
}## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
## [1] "Extracting spots"
## [1] "Preparing spot 1"
## [1] "Preparing spot 2"
## [1] "Preparing spot 3"
## [1] "Preparing spot 4"
## [1] "Preparing spot 5"
## [1] "Preparing spot 6"
## [1] "Preparing spot 7"
## [1] "Done preparation!"
count <- 0
cosine.simil_scores <- c()
for (i in c(1:4)){
for (j in c(1:4)){
if (i>j){
count <- count + 1
cosine.simil_scores[[count]] <- test_sample_and_genes(a = displace_spot2gex[[i]],b = displace_spot2gex[[j]], non_zero_markers = non_zero_markers, test_type = "t.test")
}
}
}
displace_spot2gex <- cosine.simil_scores
print(c("displace cosine correlation; sample-wise",paste(c("0vs10: ","0vs20: ","10vs20: ","0vs30: ","10vs30: ","20vs30: "),round(do.call('c',lapply(displace_spot2gex,function(X){mean(X$sample_wise)})),10))))## [1] "displace cosine correlation; sample-wise"
## [2] "0vs10: 0.9498035314"
## [3] "0vs20: 0.9184919251"
## [4] "10vs20: 0.9466096219"
## [5] "0vs30: 0.8993281351"
## [6] "10vs30: 0.9148656875"
## [7] "20vs30: 0.9470035489"
print(c("displace cosine correlation; gene-wise",paste(c("0vs10: ","0vs20: ","10vs20: ","0vs30: ","10vs30: ","20vs30: "),round(do.call('c',lapply(displace_spot2gex,function(X){mean(X$gene_wise)})),10))))## [1] "displace cosine correlation; gene-wise"
## [2] "0vs10: 0.999127193"
## [3] "0vs20: 0.9985239803"
## [4] "10vs20: 0.9991602223"
## [5] "0vs30: 0.9983144945"
## [6] "10vs30: 0.9987602255"
## [7] "20vs30: 0.9992147257"## used (Mb) gc trigger (Mb) limit (Mb) max used (Mb)
## Ncells 753686 40.3 2498607 133.5 NA 3123258 166.8
## Vcells 1372001 10.5 1535690643 11716.4 16384 1639281165 12506.8